Shared Genome Segment Analysis Documentation¶
Overview
Shared Genomic Segment (SGS) analysis is a gene mapping strategy that identifies all genomic segments shared identical-by-state (sharing without regard to inheritance) between a defined set of cases. If the length of a shared segment is significantly longer than by chance, inherited sharing is implied – theoretically, chance inherited sharing in distant relatives is extremely improbable. The software uses a dense, genome-wide map of common single nucleotide polymorphisms (SNPs), either from a genotyping platform or extracted from sequence data, to identify shared segments.
Nominal chance occurrence (nominal p-value) for shared segments is assessed empirically using gene-drop simulations to create a null distribution. First, null genotype configurations are generated by assigning haplotypes to pedigree founders according to a project specific linkage disequilibrium map. These null genotypes are then segregated through the pedigree structure to the case set via simulated Mendelian inheritance according to a genetic (recombination) map. (Gene-drops are performed independent of disease status.) The resulting genotype data in the case set are representative of chance sharing.
The SGS analysis method accounts for intra-familial heterogeneity and genome-wide multiple testing. Heterogeneity within pedigrees is accounted for in a “brute-force” fashion by iterating over all non-trivial combinations of the cases (subsets) in each pedigree. For each subset, shared segments at every position throughout the genome are identified and nominal p-values are assigned. Across subsets, an optimization procedure is performed, at every marker across the genome, to identify the segment with the most significant sharing evidence. All shared segments selected by the optimization procedure, and their respective p-values, comprise the final optimized SGS results for a pedigree.
To perform significance testing and identify segments that are unexpected by chance
(hypothesized to harbor risk loci), we derive significance thresholds to account for the
genome-wide optimization. Acknowledging that the vast majority of observed sharing across
a genome is under the null (true risk loci are a very small minority of the genome), we
use the observed optimized results (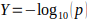 , where
, where 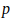 is the nominal p-value) to
model the distribution for optimized SGS results. The optimized SGS results are fitted to
a gamma distribution,
is the nominal p-value) to
model the distribution for optimized SGS results. The optimized SGS results are fitted to
a gamma distribution, 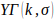 , where
, where
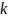 and
and
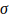 are the shape and rate
parameters. The significance threshold,
are the shape and rate
parameters. The significance threshold,
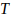 , accounts for multiple testing of
optimized segments across the genome, and is found by solving Eq. 1:
, accounts for multiple testing of
optimized segments across the genome, and is found by solving Eq. 1:
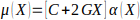 (1)
(1)
where 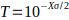 ,
,
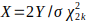 ,
,
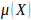 is the genome-wide false positive
rate required,
is the genome-wide false positive
rate required, 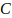 is the number of
chromosomes,
is the number of
chromosomes, 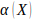 is nominal probability
of exceeding
is nominal probability
of exceeding 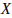 , and
, and
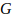 is the genome length in Morgans. A
criterion of
is the genome length in Morgans. A
criterion of 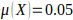 is used to define the
genome-wide significant threshold (false positive rate of 0.05 per genome), and
is used to define the
genome-wide significant threshold (false positive rate of 0.05 per genome), and
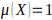 to define the genome-wide suggestive
threshold (false positive rate of 1 per genome).
to define the genome-wide suggestive
threshold (false positive rate of 1 per genome).
In general, the fitted distributions produce stable significance thresholds after 100,000-300,000 simulations. Typically, threshold determination requires 1,000-3,000 CPU hours per pedigree, increasing with the number of subsets and separating meioses between pedigree cases. Once significance thresholds are established, subset/segment combinations of potential interest can be identified and additional simulations are restricted to those combinations to gain the required p-value resolution. For these subsequent targeted simulations, a marginalized linkage disequilibrium map is used specific for the segment of interest, dramatically reducing the analysis time.
Citations
Thomas A, Camp NJ, Farnham JM, Allen-brady K, Lisa A. Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. October. 2010;72(Pt 2):279–87. Knight S, Abo RP, Abel HJ, Neklason DW, Tuohy TM, Burt RW, et al. Shared Genomic Segment Analysis: The Power to Find Rare Disease Variants. Ann Hum Genet. 2012;76(6):500–9. Waller RG, Darlington TM, Wei X, Madsen M, Thomas A, Curtin K, et al. Novel pedigree analysis implicates DNA repair and chromatin remodeling in Multiple Myeloma risk. bioRxiv [Internet]. 2017 Jan 1; Available from: http://biorxiv.org/content/early/2017/09/09/137000
Workflow
Genotypes can be generated from a high-density SNP array, or by extracting SNVs from whole-genome sequencing. CEU and GBR genotypes (unrelated individuals only) from the 1000Genomes Project are generally used as population controls. Dotted boxes represent steps done per-pedigree. Dash-dot boxes represent steps done on all subsets of cases within a pedigree. Dashed box contains step repeated for each simulation. Abbreviations: SNP – single nucleotide polymorphism; SGS – shared genomic segment; LD – linkage disequilibrium; PED – pedigree file (contains relationships and genotypes).
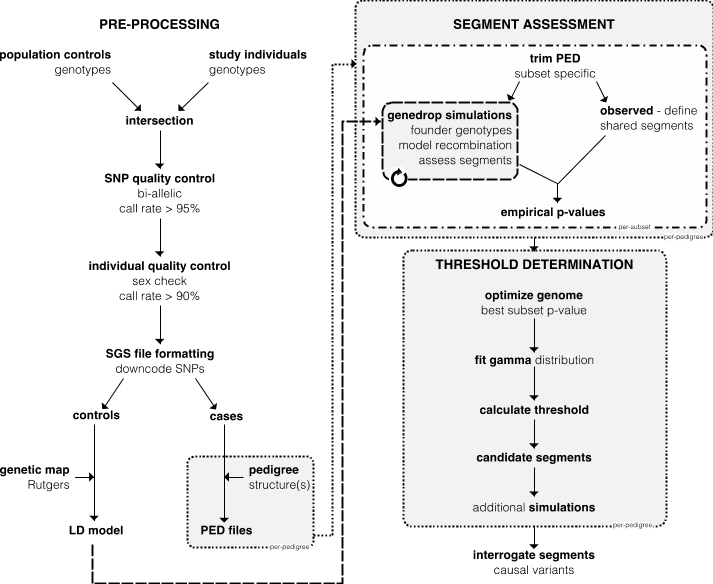
Pre-processing quality control
Prior to SGS processing the following quality control steps need to be performed on the population and individual genotypes (PLINK can be used):
- Check study individual ethnicity and select appropriate population controls
- Remove SNPs that are not bi-allelic
- Remove SNPs with < 95% call rate across all individuals
- Remove individuals that fail sex check
- Remove individuals with < 90% call rate across the genome
- Check relationship estimates against pedigree structure
- Transform QC’ed SNPs to match strand orientation of controls
Input files
- LINKAGE format PED file containing the pedigree structure and genotype data for all probands. The following PED file fields are required:
- family_id
- individual_id
- father
- mother
- 0: (place holder)
- 0: (place holder)
- 0: (place holder)
- sex: (1=male, 2=female)
- affection: (0=without genotypic data, 1=affected, with genotypic data)
- alleles: (two columns per marker)
- LINKAGE PAR file including a linkage disequilibrium model: created from the FitGMLD program (part of the available software) based on control genotypes and recombination map (such as Rutger’s)
Output files
If no database connection is specified files are written under the following structure:
A header with all arguments passed in from the command line, list of probands, meioses count between those probands, simulations completed, and version info. Then each line representing an observed segment has 9 fields:
- Chromosome number
- Starting marker index
- End marker index
- Length of region in markers ((field 3 minus field 2) + 1)
- Starting base position
- Ending base position
- Number of times the simulated data returned a region of sharing less than the length of the observed region
- Number of times the simulated data returned a region of sharing equal to the length of the observed region
- Number of times the simulated data returned a region of sharing greater than the length of the observed region
Calculates a p-value for each segment by dividing the sum of fields 8 and 9 by the sum of fields 7, 8 and 9. That is, the p-value is the number of segments defined in simulations which subsume the observed segment divided by the number of simulations.
With –confidence and –threshold in play, SGSPValue (–operation 1) will calculate rate of observations for any given segment and stop running if the specified confidence interval around this rate clears the given threshold.
Build dependencies
Java 1.8 or later. All coding dependencies except the ‘longpowerset-1.0’ and ‘jpsgcs-3.0’ will be downloaded by the build system (gradle). The former is available at https://github.com/jonroler/longpowerset and the latter is a companion of this project available at https://gitlab.com/camplab/jpsgcs
- Groovy-all-2.4.7
- jackson-annotations-2.9.2
- jackson-core-2.9.2
- jackson-databind-2.9.2
- jooq-3.13.
- jooq-meta-3.13
- jooq-codegen.jar-3.13
- jopt-simple-4.6
- jpsgcs-3.1.2
- logback-access-1.1.2
- logback-classic-1.1.2
- logback-core-1.1.2
- longpowerset-1.0
- postgresql-42.1.4
- slf4j-api-1.7.7
Build products
We now generate three separate jars, including one with the generated database code. Choosing a DBMS other than postgreSQL might require one to regenerate sgs-jooqdb.jar. A pathing jar is made with absolute paths definable at build time.